
Chronic Obstructive Pulmonary Disease: Introduction
Chronic obstructive pulmonary disease (COPD) is defined as a disease state characterized by airflow obstruction (limitation) that is not fully reversible . COPD includes emphysema, an anatomically defined condition characterized by destruction and enlargement of the lung alveoli; chronic bronchitis, a clinically defined condition with chronic cough and phlegm; and small airways disease, a condition in which small bronchioles are narrowed. COPD is present only if chronic airflow obstruction occurs; chronic bronchitis without chronic airflow obstruction is not included within COPD.
COPD is the fourth leading cause of death and affects >10 million persons in the United States. COPD is also a disease of increasing public health importance around the world. Estimates suggest that COPD will rise from the sixth to the third most common cause of death worldwide by 2020.
Risk Factors
Cigarette Smoking
By 1964, the Advisory Committee to the Surgeon General of the United States had concluded that cigarette smoking was a major risk factor for mortality from chronic bronchitis and emphysema. Subsequent longitudinal studies have shown accelerated decline in the volume of air exhaled within the first second of the forced expiratory maneuver (FEV1) in a dose-response relationship to the intensity of cigarette smoking, which is typically expressed as pack-years (average number of packs of cigarettes smoked per day multiplied by the total number of years of smoking). This dose-response relationship between reduced pulmonary function and cigarette smoking intensity accounts for the higher prevalence rates for COPD with increasing age. The historically higher rate of smoking among males is the likely explanation for the higher prevalence of COPD among males; however, the prevalence of COPD among females is increasing as the gender gap in smoking rates has diminished in the past 50 years.
Although the causal relationship between cigarette smoking and the development of COPD has been absolutely proved, there is considerable variability in the response to smoking. Although pack-years of cigarette smoking is the most highly significant predictor of FEV1 (Fig. G-1), only 15% of the variability in FEV1 is explained by pack-years. This finding suggests that additional environmental and/or genetic factors contribute to the impact of smoking on the development of airflow obstruction.
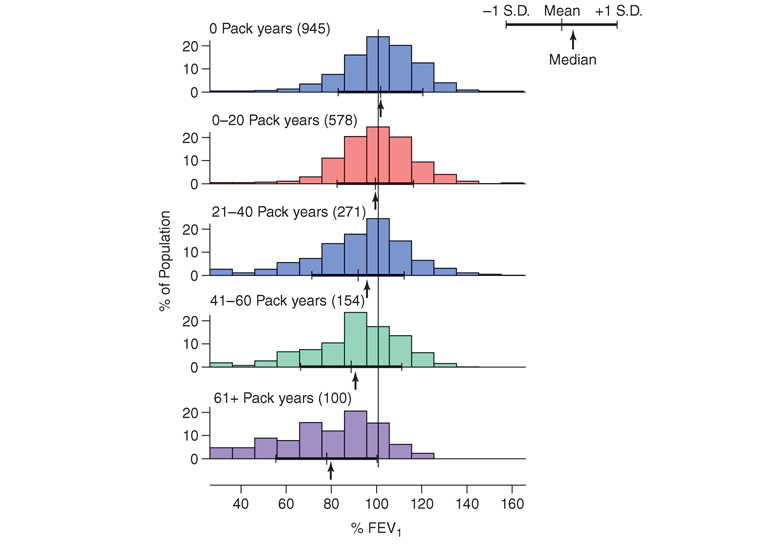
Although cigar and pipe smoking may also be associated with the development of COPD, the evidence supporting such associations is less compelling, likely related to the lower dose of inhaled tobacco by-products during cigar and pipe smoking.
Airway Responsiveness and COPD
A tendency for increased bronchoconstriction in response to a variety of exogenous stimuli, including methacholine and histamine, is one of the defining features of asthma . However, many patients with COPD also share this feature of airway hyperresponsiveness. The considerable overlap between persons with asthma and those with COPD in airway responsiveness, airflow obstruction, and pulmonary symptoms led to the formulation of the Dutch hypothesis. This suggests that asthma, chronic bronchitis, and emphysema are variations of the same basic disease, which is modulated by environmental and genetic factors to produce these pathologically distinct entities. The alternative British hypothesis contends that asthma and COPD are fundamentally different diseases: Asthma is viewed as largely an allergic phenomenon, while COPD results from smoking-related inflammation and damage. Determination of the validity of the Dutch hypothesis vs. the British hypothesis awaits identification of the genetic predisposing factors for asthma and/or COPD, as well as the interactions between these postulated genetic factors and environmental risk factors. Of note, several genes related to the proteinase-antiproteinase hypothesis have been implicated as genetic determinants for both COPD and asthma, including ADAM33 and macrophage elastase (MMP12) as described below.
Longitudinal studies that compared airway responsiveness at the beginning of the study to subsequent decline in pulmonary function have demonstrated that increased airway responsiveness is clearly a significant predictor of subsequent decline in pulmonary function. Thus, airway hyperresponsiveness is a risk factor for COPD.
Respiratory Infections
The impact of adult respiratory infections on decline in pulmonary function is controversial, but significant long-term reductions in pulmonary function are not typically seen following an episode of bronchitis or pneumonia. The impact of the effects of childhood respiratory illnesses on the subsequent development of COPD has been difficult to assess due to a lack of adequate longitudinal data. Thus, although respiratory infections are important causes of exacerbations of COPD, the association of both adult and childhood respiratory infections to the development and progression of COPD remains to be proven.
Occupational Exposures
Increased respiratory symptoms and airflow obstruction have been suggested to result from general exposure to dust and fumes at work. Several specific occupational exposures, including coal mining, gold mining, and cotton textile dust, have been suggested as risk factors for chronic airflow obstruction. Although nonsmokers in these occupations developed some reductions in FEV1, the importance of dust exposure as a risk factor for COPD, independent of cigarette smoking, is not certain for most of these exposures. However, a recent study found that coal mine dust exposure was a significant risk factor for emphysema in both smokers and nonsmokers. In most cases, the magnitude of these occupational exposures on COPD risk is likely substantially less important than the effect of cigarette smoking.
Ambient Air Pollution
Some investigators have reported increased respiratory symptoms in those living in urban compared to rural areas, which may relate to increased pollution in the urban settings. However, the relationship of air pollution to chronic airflow obstruction remains unproved. Prolonged exposure to smoke produced by biomass combustion—a common mode of cooking in some countries—also appears to be a significant risk factor for COPD among women in those countries. However, in most populations, ambient air pollution is a much less important risk factor for COPD than cigarette smoking.
Passive, or Second-Hand, Smoking Exposure
Exposure of children to maternal smoking results in significantly reduced lung growth. In utero tobacco smoke exposure also contributes to significant reductions in postnatal pulmonary function. Although passive smoke exposure has been associated with reductions in pulmonary function, the importance of this risk factor in the development of the severe pulmonary function reductions in COPD remains uncertain.
Genetic Considerations
Although cigarette smoking is the major environmental risk factor for the development of COPD, the development of airflow obstruction in smokers is highly variable. Severe alpha1 antitrypsin (alpha1AT) deficiency is a proven genetic risk factor for COPD; there is increasing evidence that other genetic determinants also exist.
Alpha1 Antitrypsin Deficiency
Many variants of the protease inhibitor (PI or SERPINA1) locus that encodes alpha1AT have been described. The common M allele is associated with normal alpha1AT levels. The S allele, associated with slightly reduced alpha1AT levels, and the Z allele, associated with markedly reduced alpha1AT levels, also occur with frequencies >1% in most white populations. Rare individuals inherit null alleles, which lead to the absence of any alpha1AT production through a heterogeneous collection of mutations. Individuals with two Z alleles or one Z and one null allele are referred to as PiZ, which is the most common form of severe alpha1AT deficiency.
Although only 1–2% of COPD patients are found to have severe alpha1AT deficiency as a contributing cause of COPD, these patients demonstrate that genetic factors can have a profound influence on the susceptibility for developing COPD. PiZ individuals often develop early-onset COPD, but the ascertainment bias in the published series of PiZ individuals—which have usually included many PiZ subjects who were tested for alpha1AT deficiency because they had COPD—means that the fraction of PiZ individuals who will develop COPD and the age-of-onset distribution for the development of COPD in PiZ subjects remain unknown. Approximately 1 in 3000 individuals in the United States inherits severe alpha1AT deficiency, but only a small minority of these individuals has been recognized. The clinical laboratory test used most frequently to screen for alpha1AT deficiency is measurement of the immunologic level of alpha1AT in serum (see “Laboratory Findings”).
A significant percentage of the variability in pulmonary function among PiZ individuals is explained by cigarette smoking; cigarette smokers with severe alpha1AT deficiency are more likely to develop COPD at early ages. However, the development of COPD in PiZ subjects, even among current or ex-smokers, is not absolute. Among PiZ nonsmokers, impressive variability has been noted in the development of airflow obstruction. Asthma and male gender also appear to increase the risk of COPD in PiZ subjects. Other genetic and/or environmental factors likely contribute to this variability.
Specific treatment in the form of alpha1AT augmentation therapy is available for severe alpha1AT deficiency as a weekly IV infusion (see “Treatment,” below).
The risk of lung disease in heterozygous PiMZ individuals, who have intermediate serum levels of alpha1AT (~60% of PiMM levels), is controversial. Although previous general population surveys have not typically shown increased rates of airflow obstruction in PiMZ compared to PiMM individuals, case-control studies that compared COPD patients to control subjects have usually found an excess of PiMZ genotypes in the COPD patient group. Several recent large population studies have suggested that PiMZ subjects are at slightly increased risk for the development of airflow obstruction, but it remains unclear if all PiMZ subjects are at slightly increased risk for COPD or if a subset of PiMZ subjects are at substantially increased risk for COPD due to other genetic or environmental factors.
Other Genetic Risk Factors
Studies of pulmonary function measurements performed in general population samples have suggested that genetic factors other than PI type influence variation in pulmonary function. Familial aggregation of airflow obstruction within families of COPD patients has also been demonstrated.
Association studies have compared the distribution of variants in candidate genes hypothesized to be involved in the development of COPD in COPD patients and control subjects. However, the results have been quite inconsistent, often due to underpowered studies. However, a recent association study comprising 8300 patients and 7 separate cohorts found that a minor allele SNP of MMP12 (rs2276109) associated with decreased MMP-12 expression has a positive effect on lung function in children with asthma and in adult smokers. Recent genome-wide association studies have identified several COPD loci, including a region near the hedgehog interacting protein (HHIP) gene on chromosome 4 and a cluster of genes on chromosome 15 (including components of the nicotinic acetylcholine receptor) that likely contain COPD susceptibility determinants, but the specific genetic determinants in those regions have yet to be definitively identified.
Natural History
The effects of cigarette smoking on pulmonary function appear to depend on the intensity of smoking exposure, the timing of smoking exposure during growth, and the baseline lung function of the individual; other environmental factors may have similar effects. Most individuals follow a steady trajectory of increasing pulmonary function with growth during childhood and adolescence, followed by a gradual decline with aging. Individuals appear to track in their quartile of pulmonary function based upon environmental and genetic factors that put them on different tracks. The risk of eventual mortality from COPD is closely associated with reduced levels of FEV1. A graphic depiction of the natural history of COPD is shown as a function of the influences on tracking curves of FEV1 in Fig. G-2. Death or disability from COPD can result from a normal rate of decline after a reduced growth phase (curve C), an early initiation of pulmonary function decline after normal growth (curve B), or an accelerated decline after normal growth (curve D). The rate of decline in pulmonary function can be modified by changing environmental exposures (i.e., quitting smoking), with smoking cessation at an earlier age providing a more beneficial effect than smoking cessation after marked reductions in pulmonary function have already developed. Genetic factors likely contribute to the level of pulmonary function achieved during growth and to the rate of decline in response to smoking and potentially to other environmental factors as well.
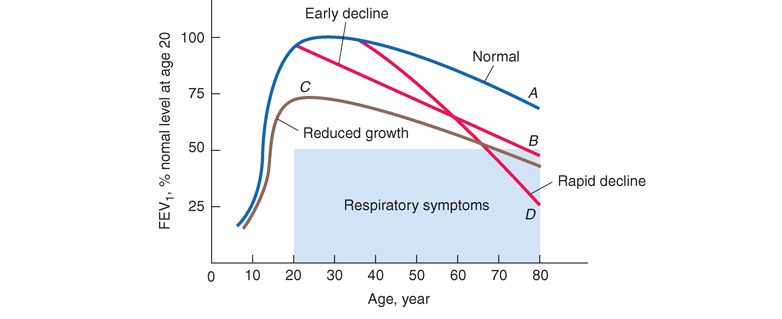
Pathophysiology
Persistent reduction in forced expiratory flow rates is the most typical finding in COPD. Increases in the residual volume and the residual volume/total lung capacity ratio, nonuniform distribution of ventilation, and ventilation-perfusion mismatching also occur.
Airflow Obstruction
Airflow limitation, also known as airflow obstruction, is typically determined by spirometry, which involves forced expiratory maneuvers after the subject has inhaled to total lung capacity. Key parameters obtained from spirometry include FEV1 and the total volume of air exhaled during the entire spirometric maneuver [forced vital capacity (FVC)]. Patients with airflow obstruction related to COPD have a chronically reduced ratio of FEV1/FVC. In contrast to asthma, the reduced FEV1 in COPD seldom shows large responses to inhaled bronchodilators, although improvements up to 15% are common. Asthma patients can also develop chronic (not fully reversible) airflow obstruction.
Airflow during forced exhalation is the result of the balance between the elastic recoil of the lungs promoting flow and the resistance of the airways limiting flow. In normal lungs, as well as in lungs affected by COPD, maximal expiratory flow diminishes as the lungs empty because the lung parenchyma provides progressively less elastic recoil and because the cross-sectional area of the airways falls, raising the resistance to airflow. The decrease in flow coincident with decreased lung volume is readily apparent on the expiratory limb of a flow-volume curve. In the early stages of COPD, the abnormality in airflow is only evident at lung volumes at or below the functional residual capacity (closer to residual volume), appearing as a scooped-out lower part of the descending limb of the flow-volume curve. In more advanced disease the entire curve has decreased expiratory flow compared to normal.
Hyperinflation
Lung volumes are also routinely assessed in pulmonary function testing. In COPD there is often “air trapping” (increased residual volume and increased ratio of residual volume to total lung capacity) and progressive hyperinflation (increased total lung capacity) late in the disease. Hyperinflation of the thorax during tidal breathing preserves maximum expiratory airflow, because as lung volume increases, elastic recoil pressure increases, and airways enlarge so that airway resistance decreases.
Despite compensating for airway obstruction, hyperinflation can push the diaphragm into a flattened position with a number of adverse effects. First, by decreasing the zone of apposition between the diaphragm and the abdominal wall, positive abdominal pressure during inspiration is not applied as effectively to the chest wall, hindering rib cage movement and impairing inspiration. Second, because the muscle fibers of the flattened diaphragm are shorter than those of a more normally curved diaphragm, they are less capable of generating inspiratory pressures than normal. Third, the flattened diaphragm (with increased radius of curvature, r) must generate greater tension (t) to develop the transpulmonary pressure (p) required to produce tidal breathing. This follows from Laplace’s law, p = 2t/r. Also, because the thoracic cage is distended beyond its normal resting volume, during tidal breathing the inspiratory muscles must do work to overcome the resistance of the thoracic cage to further inflation instead of gaining the normal assistance from the chest wall recoiling outward toward its resting volume.
Gas Exchange
Although there is considerable variability in the relationships between the FEV1 and other physiologic abnormalities in COPD, certain generalizations may be made. The PaO2 usually remains near normal until the FEV1 is decreased to ~50% of predicted, and even much lower FEV1 values can be associated with a normal PaO2, at least at rest. An elevation of arterial level of carbon dioxide (PaCO2) is not expected until the FEV1 is <25% of predicted and even then may not occur. Pulmonary hypertension severe enough to cause cor pulmonale and right ventricular failure due to COPD typically occurs in individuals who have marked decreases in FEV1 (<25% of predicted) and chronic hypoxemia (PaO2 <55 mmHg); however, recent evidence suggests that some patients will develop significant pulmonary hypertension independent of COPD severity .
Nonuniform ventilation and ventilation-perfusion mismatching are characteristic of COPD, reflecting the heterogeneous nature of the disease process within the airways and lung parenchyma. Physiologic studies are consistent with multiple parenchymal compartments having different rates of ventilation due to regional differences in compliance and airway resistance. Ventilation-perfusion mismatching accounts for essentially all of the reduction in PaO2 that occurs in COPD; shunting is minimal. This finding explains the effectiveness of modest elevations of inspired oxygen in treating hypoxemia due to COPD and therefore the need to consider problems other than COPD when hypoxemia is difficult to correct with modest levels of supplemental oxygen in the patient with COPD.
Pathology
Cigarette smoke exposure may affect the large airways, small airways (≤2 mm diameter), and alveoli. Changes in large airways cause cough and sputum, while changes in small airways and alveoli are responsible for physiologic alterations. Emphysema and small airway pathology are both present in most persons with COPD; however, they do not appear to be mechanistically related to each other, and their relative contributions to obstruction vary from one person to another.
Large Airway
Cigarette smoking often results in mucous gland enlargement and goblet cell hyperplasia leading to cough and mucus production that define chronic bronchitis, but these abnormalities are not related to airflow limitation. Goblet cells not only increase in number but in extent through the bronchial tree. Bronchi also undergo squamous metaplasia, predisposing to carcinogenesis and disrupting mucociliary clearance. Although not as prominent as in asthma, patients may have smooth-muscle hypertrophy and bronchial hyperreactivity leading to airflow limitation. Neutrophil influx has been associated with purulent sputum of upper respiratory tract infections. Independent of its proteolytic activity, neutrophil elastase is among the most potent secretagogues identified.
Small Airways
The major site of increased resistance in most individuals with COPD is in airways ≤2 mm diameter. Characteristic cellular changes include goblet cell metaplasia, with these mucus-secreting cells replacing surfactant-secreting Clara cells. Infiltration of mononuclear phagocytes is also prominent. Smooth-muscle hypertrophy may also be present. These abnormalities may cause luminal narrowing by fibrosis, excess mucus, edema, and cellular infiltration. Reduced surfactant may increase surface tension at the air-tissue interface, predisposing to airway narrowing or collapse. Respiratory bronchiolitis with mononuclear inflammatory cells collecting in distal airway tissues may cause proteolytic destruction of elastic fibers in the respiratory bronchioles and alveolar ducts where the fibers are concentrated as rings around alveolar entrances.
Because small airway patency is maintained by the surrounding lung parenchyma that provides radial traction on bronchioles at points of attachment to alveolar septa, loss of bronchiolar attachments as a result of extracellular matrix destruction may cause airway distortion and narrowing in COPD.
Lung Parenchyma
Emphysema is characterized by destruction of gas-exchanging air spaces, i.e., the respiratory bronchioles, alveolar ducts, and alveoli. Their walls become perforated and later obliterated with coalescence of small distinct air spaces into abnormal and much larger air spaces. Macrophages accumulate in respiratory bronchioles of essentially all young smokers. Bronchoalveolar lavage fluid from such individuals contains roughly five times as many macrophages as lavage from nonsmokers. In smokers’ lavage fluid, macrophages comprise >95% of the total cell count, and neutrophils, nearly absent in nonsmokers’ lavage, account for 1–2% of the cells. T lymphocytes, particularly CD8+ cells, are also increased in the alveolar space of smokers.
Emphysema is classified into distinct pathologic types, the most important being centriacinar and panacinar. Centriacinar emphysema, the type most frequently associated with cigarette smoking, is characterized by enlarged air spaces found (initially) in association with respiratory bronchioles. Centriacinar emphysema is usually most prominent in the upper lobes and superior segments of lower lobes and is often quite focal. Panacinar emphysema refers to abnormally large air spaces evenly distributed within and across acinar units. Panacinar emphysema is usually observed in patients with alpha1AT deficiency, which has a predilection for the lower lobes. Distinctions between centriacinar and panacinar emphysema are interesting and may ultimately be shown to have different mechanisms of pathogenesis. However, garden-variety smoking-related emphysema is usually mixed, particularly in advanced cases, and these pathologic classifications are not helpful in the care of patients with COPD.
Pathogenesis
Airflow limitation, the major physiologic change in COPD, can result from both small airway obstruction and emphysema, as discussed above. Pathologic findings that can contribute to small airway obstruction are described above, but their relative importance is unknown. Fibrosis surrounding the small airways appears to be a significant contributor. Mechanisms leading to collagen accumulation around the airways in the face of increased collagenase activity remain an enigma. Although seemingly counterintuitive, there are several potential mechanisms whereby a proteinase can predispose to fibrosis, including proteolytic activation of transforming growth factor Beta (TGF-Beta). Largely due to greater similarity of animal air spaces than airways to humans, we know much more about mechanisms involved in emphysema than small airway obstruction.
The dominant paradigm of the pathogenesis of emphysema comprises four interrelated events (Fig. G-3): (1) Chronic exposure to cigarette smoke may lead to inflammatory cell recruitment within the terminal air spaces of the lung. (2) These inflammatory cells release elastolytic proteinases that damage the extracellular matrix of the lung. (3) Structural cell death results from oxidant stress and loss of matrix-cell attachment. (4) Ineffective repair of elastin and other extracellular matrix components result in air space enlargement that defines pulmonary emphysema.

The Elastase:Antielastase Hypothesis
Elastin, the principal component of elastic fibers, is a highly stable component of the extracellular matrix that is critical to the integrity of the lung. The elastase:antielastase hypothesis proposed in the mid-1960s states that the balance of elastin-degrading enzymes and their inhibitors determines the susceptibility of the lung to destruction resulting in air space enlargement. This hypothesis was based on the clinical observation that patients with genetic deficiency in alpha1AT, the inhibitor of the serine proteinase neutrophil elastase, were at increased risk of emphysema, and that instillation of elastases, including neutrophil elastase, to experimental animals results in emphysema. The elastase:antielastase hypothesis remains a prevailing mechanism for the development of emphysema. However, a complex network of immune and inflammatory cells and additional proteinases that contribute to emphysema have subsequently been identified.
Inflammation and Extracellular Matrix Proteolysis
Macrophages patrol the lower air space under normal conditions. Upon exposure to oxidants from cigarette smoke, macrophages become activated, producing proteinases and chemokines that attract other inflammatory cells. One mechanism of macrophage activation occurs via oxidant-induced inactivation of histone deacetylase-2, shifting the balance toward acetylated or loose chromatin, exposing nuclear factor kB sites and resulting in transcription of matrix metalloproteinases, proinflammatory cytokines such as interleukin 8 (IL-8), and tumor necrosis factor alpha (TNF-alpha); this leads to neutrophil recruitment. CD8+ T cells are also recruited in response to cigarette smoke and release interferon inducible protein-10 (IP-10, CXCL-7) that in turn leads to macrophage production of macrophage elastase [matrix metalloproteinase-12 (MMP-12)]. Matrix metalloproteinases and serine proteinases, most notably neutrophil elastase, work together by degrading the inhibitor of the other, leading to lung destruction. Proteolytic cleavage products of elastin also serve as a macrophage chemokine, fueling this destructive positive feedback loop.
Autoimmune mechanisms have recently been identified in COPD and may promote the progression of disease. Increased B cells and lymphoid follicles are present in patients, particularly those with advanced disease. Antibodies have been found against elastin fragments, as well; IgG autoantibodies with avidity for pulmonary epithelium and the potential to mediate cytotoxicity have been detected.
Concomitant cigarette smoke–induced loss of cilia in the airway epithelium and impaired macrophage phagocytosis predispose to bacterial infection with neutrophilia. In end-stage lung disease, long after smoking cessation there remains an exuberant inflammatory response, suggesting that mechanisms of cigarette smoke–induced inflammation that initiate the disease differ from mechanisms that sustain inflammation after smoking cessation.
Cell Death
Air space enlargement with loss of alveolar units obviously requires disappearance of both extracellular matrix and cells. Cell death can occur from increased oxidant stress both directly from cigarette smoke and from inflammation. Animal models have used endothelial and epithelial cell death as a means to generate transient air space enlargement. Uptake of apoptotic cells by macrophages results in production of growth factors and dampens inflammation, promoting lung repair. Cigarette smoke impairs macrophage uptake of apoptotic cells, limiting repair.
Ineffective Repair
The ability of the adult lung to repair damaged alveoli appears limited. It is unlikely that the process of septation that is responsible for alveologenesis during lung development can be reinitiated. The capacity of stem cells to repopulate the lung is under active investigation. It appears difficult for an adult human to completely restore an appropriate extracellular matrix, particularly functional elastic fibers.
Clinical Presentation
History
The three most common symptoms in COPD are cough, sputum production, and exertional dyspnea. Many patients have such symptoms for months or years before seeking medical attention. Although the development of airflow obstruction is a gradual process, many patients date the onset of their disease to an acute illness or exacerbation. A careful history, however, usually reveals the presence of symptoms prior to the acute exacerbation. The development of exertional dyspnea, often described as increased effort to breathe, heaviness, air hunger, or gasping, can be insidious. It is best elicited by a careful history focused on typical physical activities and how the patient’s ability to perform them has changed. Activities involving significant arm work, particularly at or above shoulder level, are particularly difficult for patients with COPD. Conversely, activities that allow the patient to brace the arms and use accessory muscles of respiration are better tolerated. Examples of such activities include pushing a shopping cart, walking on a treadmill, or pushing a wheelchair. As COPD advances, the principal feature is worsening dyspnea on exertion with increasing intrusion on the ability to perform vocational or avocational activities. In the most advanced stages, patients are breathless doing simple activities of daily living.
Accompanying worsening airflow obstruction is an increased frequency of exacerbations (described below). Patients may also develop resting hypoxemia and require institution of supplemental oxygen.
Physical Findings
In the early stages of COPD, patients usually have an entirely normal physical examination. Current smokers may have signs of active smoking, including an odor of smoke or nicotine staining of fingernails. In patients with more severe disease, the physical examination is notable for a prolonged expiratory phase and may include expiratory wheezing. In addition, signs of hyperinflation include a barrel chest and enlarged lung volumes with poor diaphragmatic excursion as assessed by percussion. Patients with severe airflow obstruction may also exhibit use of accessory muscles of respiration, sitting in the characteristic “tripod” position to facilitate the actions of the sternocleidomastoid, scalene, and intercostal muscles. Patients may develop cyanosis, visible in the lips and nail beds.
Although traditional teaching is that patients with predominant emphysema, termed “pink puffers,” are thin and noncyanotic at rest and have prominent use of accessory muscles, and patients with chronic bronchitis are more likely to be heavy and cyanotic (“blue bloaters”), current evidence demonstrates that most patients have elements of both bronchitis and emphysema and that the physical examination does not reliably differentiate the two entities.
Advanced disease may be accompanied by systemic wasting, with significant weight loss, bitemporal wasting, and diffuse loss of subcutaneous adipose tissue. This syndrome has been associated with both inadequate oral intake and elevated levels of inflammatory cytokines (TNF-alpha). Such wasting is an independent poor prognostic factor in COPD. Some patients with advanced disease have paradoxical inward movement of the rib cage with inspiration (Hoover’s sign), the result of alteration of the vector of diaphragmatic contraction on the rib cage as a result of chronic hyperinflation.
Signs of overt right heart failure, termed cor pulmonale, are relatively infrequent since the advent of supplemental oxygen therapy.
Clubbing of the digits is not a sign of COPD, and its presence should alert the clinician to initiate an investigation for causes of clubbing. In this population, the development of lung cancer is the most likely explanation for newly developed clubbing.
Laboratory Findings
The hallmark of COPD is airflow obstruction (discussed above). Pulmonary function testing shows airflow obstruction with a reduction in FEV1 and FEV1/FVC . With worsening disease severity, lung volumes may increase, resulting in an increase in total lung capacity, functional residual capacity, and residual volume. In patients with emphysema, the diffusing capacity may be reduced, reflecting the lung parenchymal destruction characteristic of the disease. The degree of airflow obstruction is an important prognostic factor in COPD and is the basis for the Global Initiative for Lung Disease (GOLD) redundant classification (Table G-1). More recently it has been shown that a multifactorial index incorporating airflow obstruction, exercise performance, dyspnea, and body mass index is a better predictor of mortality rate than pulmonary function alone.
Table 260-1 Gold Criteria for COPD Severity
| GOLD Stage | Severity | Symptoms | Spirometry |
| 0 | At Risk | Chronic cough, sputum production | Normal |
| I | Mild | With or without chronic cough or sputum production | FEV1/FVC <0.7 and FEV1 ≥80% predicted |
| II | Moderate | With or without chronic cough or sputum production | FEV1/FVC <0.7 andFEV1 ≥50% but <80% predicted |
| III | Severe | With or without chronic cough or sputum production | FEV1/FVC <0.7 and FEV1≥30% but <50% predicted |
| IV | Very Severe | With or without chronic cough or sputum production | FEV1/FVC <0.7 and FEV1 <30% predicted |
Arterial blood gases and oximetry may demonstrate resting or exertional hypoxemia. Arterial blood gases provide additional information about alveolar ventilation and acid-base status by measuring arterial PCO2 and pH. The change in pH with PCO2 is 0.08 units/10 mmHg acutely and 0.03 units/10 mmHg in the chronic state. Knowledge of the arterial pH therefore allows the classification of ventilatory failure, defined as PCO2 >45 mmHg, into acute or chronic conditions. The arterial blood gas is an important component of the evaluation of patients presenting with symptoms of an exacerbation. An elevated hematocrit suggests the presence of chronic hypoxemia, as does the presence of signs of right ventricular hypertrophy.
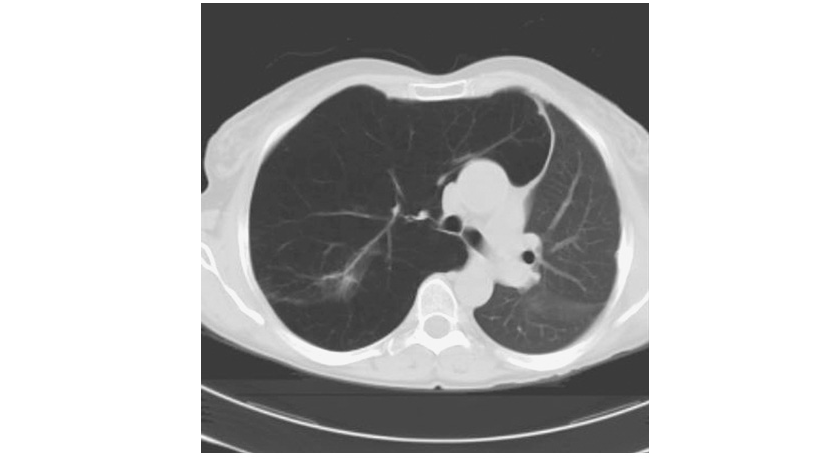
Radiographic studies may assist in the classification of the type of COPD. Obvious bullae, paucity of parenchymal markings, or hyperlucency suggests the presence of emphysema. Increased lung volumes and flattening of the diaphragm suggest hyperinflation but do not provide information about chronicity of the changes. Computed tomography (CT) scan is the current definitive test for establishing the presence or absence of emphysema in living subjects (Fig. G-4) and (Fig. G-5). From a practical perspective, the CT scan does little to influence therapy of COPD except in those individuals considering surgical therapy for their disease (described below).
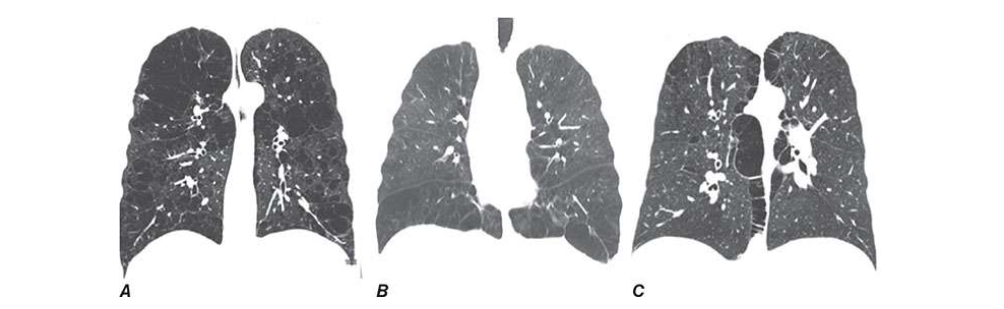
severe upper lobe involvement in a 68-year-old man with a 70-pack-year smoking
history but forced expiratory volume in 1 s (FEV1) 81% predicted (GOLD
spirometry grade 1). B. Panlobular emphysema with diffuse loss of lung
parenchymal detail predominantly in the lower lobes in a 64-year-old man with
severe α1 antitrypsin (α1AT) deficiency. C. Paraseptal emphysema with marked
airway inflammation in a 52-year-old woman with a 37-pack-year smoking history
and FEV1 40% predicted.
Recent guidelines have suggested testing for alpha1AT deficiency in all subjects with COPD or asthma with chronic airflow obstruction. Measurement of the serum alpha1AT level is a reasonable initial test. For subjects with low alpha1AT levels, the definitive diagnosis of alpha1AT deficiency requires protease inhibitor (PI) type determination. This is typically performed by isoelectric focusing of serum, which reflects the genotype at the PI locus for the common alleles and many of the rare PI alleles as well. Molecular genotyping of DNA can be performed for the common PI alleles (M, S, and Z).
Treatment: Chronic Obstructive Pulmonary Disease
Stable Phase COPD
Only three interventions—smoking cessation, oxygen therapy in chronically hypoxemic patients, and lung volume reduction surgery in selected patients with emphysema—have been demonstrated to influence the natural history of patients with COPD. There is currently suggestive, but not definitive, evidence that the use of inhaled glucocorticoids may alter mortality rate (but not lung function). All other current therapies are directed at improving symptoms and decreasing the frequency and severity of exacerbations. The institution of these therapies should involve an assessment of symptoms, potential risks, costs, and benefits of therapy. This should be followed by an assessment of response to therapy, and a decision should be made whether or not to continue treatment.
Pharmacotherapy
Smoking Cessation
It has been shown that middle-aged smokers who were able to successfully stop smoking experienced a significant improvement in the rate of decline in pulmonary function, returning to annual changes similar to that of nonsmoking patients. Thus, all patients with COPD should be strongly urged to quit and educated about the benefits of quitting. An emerging body of evidence demonstrates that combining pharmacotherapy with traditional supportive approaches considerably enhances the chances of successful smoking cessation. There are three principal pharmacologic approaches to the problem: bupropion, originally developed as an antidepressant medication; nicotine replacement therapy available as gum, transdermal patch, inhaler, and nasal spray; and varenicline, a nicotinic acid receptor agonist/antagonist. Current recommendations from the U.S. Surgeon General are that all adult, nonpregnant smokers considering quitting be offered pharmacotherapy, in the absence of any contraindication to treatment.
Bronchodilators
In general, bronchodilators are used for symptomatic benefit in patients with COPD. The inhaled route is preferred for medication delivery as the incidence of side effects is lower than that seen with the use of parenteral medication delivery.
Anticholinergic Agents
Ipratropium bromide improves symptoms and produces acute improvement in FEV1. Tiotropium, a long-acting anticholinergic, has been shown to improve symptoms and reduce exacerbations. Studies of both ipratropium and tiotropium have failed to demonstrate that either influences the rate of decline in FEV1. In a large randomized clinical trial, there was a trend toward reduced mortality rate in the tiotropium-treated patients that approached, but did not reach, statistical significance. Side effects are minor, and a trial of inhaled anticholinergics is recommended in symptomatic patients with COPD.
Beta Agonists
These provide symptomatic benefit. The main side effects are tremor and tachycardia. Long-acting inhaled Beta agonists, such as salmeterol, have benefits comparable to ipratropium bromide. Their use is more convenient than short-acting agents. The addition of a Beta agonist to inhaled anticholinergic therapy has been demonstrated to provide incremental benefit. A recent report in asthma suggests that those patients, particularly African Americans, using a long-acting Beta agonist without concomitant inhaled corticosteroids have an increased risk of deaths from respiratory causes. The applicability of these data to patients with COPD is unclear.
Inhaled Glucocorticoids
Although a recent trial demonstrated an apparent benefit from the regular use of inhaled glucocorticoids on the rate of decline of lung function, a number of other well-designed randomized trials have not. Patients studied included those with mild to severe airflow obstruction and current and ex-smokers. Patients with significant acute response to inhaled Beta agonists were excluded from many of these trials, which may impact the generalizability of the findings. Their use has been associated with increased rates of oropharyngeal candidiasis and an increased rate of loss of bone density. Available data suggest that inhaled glucocorticoids reduce exacerbation frequency by ~25%. The impact of inhaled corticosteroids on mortality rates in COPD is controversial. A meta-analysis and several retrospective studies suggest a mortality benefit, but in a recently published randomized trial, differences in mortality rate approached, but did not reach, conventional criteria for statistical significance. A trial of inhaled glucocorticoids should be considered in patients with frequent exacerbations, defined as two or more per year, and in patients who demonstrate a significant amount of acute reversibility in response to inhaled bronchodilators.
The chronic use of oral glucocorticoids for treatment of COPD is not recommended because of an unfavorable benefit/risk ratio. The chronic use of oral glucocorticoids is associated with significant side effects, including osteoporosis, weight gain, cataracts, glucose intolerance, and increased risk of infection. A recent study demonstrated that patients tapered off chronic low-dose prednisone (~10 mg/d) did not experience any adverse effect on the frequency of exacerbations, health-related quality of life, or lung function. On average, patients lost ~4.5 kg (~10 lb) when steroids were withdrawn.
Theophylline produces modest improvements in expiratory flow rates and vital capacity and a slight improvement in arterial oxygen and carbon dioxide levels in patients with moderate to severe COPD. Nausea is a common side effect; tachycardia and tremor have also been reported. Monitoring of blood theophylline levels is typically required to minimize toxicity.
Oxygen
Supplemental O2 is the only pharmacologic therapy demonstrated to unequivocally decrease mortality rates in patients with COPD. For patients with resting hypoxemia (resting O2 saturation ≤88% or <90% with signs of pulmonary hypertension or right heart failure), the use of O2 has been demonstrated to have a significant impact on mortality rate. Patients meeting these criteria should be on continual oxygen supplementation, as the mortality benefit is proportional to the number of hours/day oxygen is used. Various delivery systems are available, including portable systems that patients may carry to allow mobility outside the home.
Supplemental O2 is commonly prescribed for patients with exertional hypoxemia or nocturnal hypoxemia. Although the rationale for supplemental O2 in these settings is physiologically sound, the benefits of such therapy are not well substantiated.
Other Agents
N-acetyl cysteine has been used in patients with COPD for both its mucolytic and antioxidant properties. A prospective trial failed to find any benefit with respect to decline in lung function or prevention of exacerbations. Specific treatment in the form of IV alpha1AT augmentation therapy is available for individuals with severe alpha1AT deficiency. Despite sterilization procedures for these blood-derived products and the absence of reported cases of viral infection from therapy, some physicians recommend hepatitis B vaccination prior to starting augmentation therapy. Although biochemical efficacy of alpha1AT augmentation therapy has been shown, a randomized controlled trial of alpha1AT augmentation therapy has not definitively established the efficacy of augmentation therapy in reducing decline of pulmonary function. Eligibility for alpha1AT augmentation therapy requires a serum alpha1AT level <11 microM (approximately 50 mg/dL). Typically, PiZ individuals will qualify, although other rare types associated with severe deficiency (e.g., null-null) are also eligible. Since only a fraction of individuals with severe alpha1AT deficiency will develop COPD, alpha1AT augmentation therapy is not recommended for severely alpha1AT-deficient persons with normal pulmonary function and a normal chest CT scan.
Nonpharmacologic Therapies
General Medical Care
Patients with COPD should receive the influenza vaccine annually. Polyvalent pneumococcal vaccine is also recommended, although proof of efficacy in this patient population is not definitive.
Pulmonary Rehabilitation
This refers to a treatment program that incorporates education and cardiovascular conditioning. In COPD, pulmonary rehabilitation has been demonstrated to improve health-related quality of life, dyspnea, and exercise capacity. It has also been shown to reduce rates of hospitalization over a 6- to 12-month period.
Lung Volume Reduction Surgery (LVRS)
Surgery to reduce the volume of lung in patients with emphysema was first introduced with minimal success in the 1950s and was reintroduced in the 1990s. Patients are excluded if they have significant pleural disease, a pulmonary artery systolic pressure >45 mmHg, extreme deconditioning, congestive heart failure, or other severe comorbid conditions. Patients with an FEV1 <20% of predicted and either diffusely distributed emphysema on CT scan or diffusing capacity of lung for carbon monoxide (DLCO) <20% of predicted have an increased mortality rate after the procedure and thus are not candidates for LVRS.
The National Emphysema Treatment trial demonstrated that LVRS offers both a mortality benefit and a symptomatic benefit in certain patients with emphysema. The anatomic distribution of emphysema and postrehabilitation exercise capacity are important prognostic characteristics. Patients with upper lobe–predominant emphysema and a low postrehabilitation exercise capacity are most likely to benefit from LVRS.
Lung Transplantation
COPD is currently the second leading indication for lung transplantation (Fig. G-4). Current recommendations are that candidates for lung transplantation should be <65 years; have severe disability despite maximal medical therapy; and be free of comorbid conditions such as liver, renal, or cardiac disease. In contrast to LVRS, the anatomic distribution of emphysema and the presence of pulmonary hypertension are not contraindications to lung transplantation. Unresolved issues concerning lung transplantation and COPD include whether single- or double-lung transplant is the preferred procedure.
Exacerbations of COPD
Exacerbations are a prominent feature of the natural history of COPD. Exacerbations are episodes of increased dyspnea and cough and change in the amount and character of sputum. They may or may not be accompanied by other signs of illness, including fever, myalgias, and sore throat. Self-reported health-related quality of life correlates with frequency of exacerbations more closely than it does with the degree of airflow obstruction. Economic analyses have shown that >70% of COPD-related health care expenditures go to emergency department visits and hospital care; this translates to >$10 billion annually in the United States. The frequency of exacerbations increases as airflow obstruction increases; patients with moderate to severe airflow obstruction [GOLD stages III, IV (Table G-1)] on average have one to three episodes per year. However, some individuals with very severe airflow obstruction do not have frequent exacerbations; the history of prior exacerbations is a strong predictor of future exacerbations.
The approach to the patient experiencing an exacerbation includes an assessment of the severity of the patient’s illness, both acute and chronic components; an attempt to identify the precipitant of the exacerbation; and the institution of therapy.
Precipitating Causes and Strategies to Reduce Frequency of Exacerbations
A variety of stimuli may result in the final common pathway of airway inflammation and increased symptoms that are characteristic of COPD exacerbations. Bacterial infections play a role in many, but by no means all, episodes. Viral respiratory infections are present in approximately one-third of COPD exacerbations. In a significant minority of instances (20–35%), no specific precipitant can be identified.
Despite the frequent implication of bacterial infection, chronic suppressive or “rotating” antibiotics are not beneficial in patients with COPD. This is in contrast to their efficacy in patients with bronchiectasis due to cystic fibrosis, in whom suppressive antibiotics have been shown to reduce frequency of hospital admissions.
The role of pharmacotherapy in reducing exacerbation frequency is less well studied. Chronic oral glucocorticoids are not recommended for this purpose. Inhaled glucocorticoids reduce the frequency of exacerbations by 25–30% in most analyses. The use of inhaled glucocorticoids should be considered in patients with frequent exacerbations or those who have an asthmatic component, i.e., significant reversibility on pulmonary function testing or marked symptomatic improvement after inhaled bronchodilators. Similar magnitudes of reduction have been reported for anticholinergic and long-acting beta-agonist therapy. The influenza vaccine has been shown to reduce exacerbation rates in patients with COPD.
Patient Assessment
An attempt should be made to establish the severity of the exacerbation as well as the severity of preexisting COPD. The more severe either of these two components, the more likely that the patient will require hospital admission. The history should include quantification of the degree of dyspnea by asking about breathlessness during activities of daily living and typical activities for the patient. The patient should be asked about fever; change in character of sputum; any ill contacts; and associated symptoms such as nausea, vomiting, diarrhea, myalgias, and chills. Inquiring about the frequency and severity of prior exacerbations can provide important information.
The physical examination should incorporate an assessment of the degree of distress of the patient. Specific attention should be focused on tachycardia, tachypnea, use of accessory muscles, signs of perioral or peripheral cyanosis, the ability to speak in complete sentences, and the patient’s mental status. The chest examination should establish the presence or absence of focal findings, degree of air movement, presence or absence of wheezing, asymmetry in the chest examination (suggesting large airway obstruction or pneumothorax mimicking an exacerbation), and the presence or absence of paradoxical motion of the abdominal wall.
Patients with severe underlying COPD, who are in moderate or severe distress or those with focal findings should have a chest x-ray. Approximately 25% of x-rays in this clinical situation will be abnormal, with the most frequent findings being pneumonia and congestive heart failure. Patients with advanced COPD, those with a history of hypercarbia, those with mental status changes (confusion, sleepiness), or those in significant distress should have an arterial blood-gas measurement. The presence of hypercarbia, defined as a PCO2 >45 mmHg, has important implications for treatment (discussed below). In contrast to its utility in the management of exacerbations of asthma, measurement of pulmonary function has not been demonstrated to be helpful in the diagnosis or management of exacerbations of COPD.
There are no definitive guidelines concerning the need for inpatient treatment of exacerbations. Patients with respiratory acidosis and hypercarbia, significant hypoxemia, or severe underlying disease or those whose living situation is not conducive to careful observation and the delivery of prescribed treatment should be admitted to the hospital.
Acute Exacerbations
Bronchodilators
Typically, patients are treated with an inhaled Beta agonist, often with the addition of an anticholinergic agent. These may be administered separately or together, and the frequency of administration depends on the severity of the exacerbation. Patients are often treated initially with nebulized therapy, as such treatment is often easier to administer in older patients or to those in respiratory distress. It has been shown, however, that conversion to metered-dose inhalers is effective when accompanied by education and training of patients and staff. This approach has significant economic benefits and also allows an easier transition to outpatient care. The addition of methylxanthines (such as theophylline) to this regimen can be considered, although convincing proof of its efficacy is lacking. If added, serum levels should be monitored in an attempt to minimize toxicity.
Antibiotics
Patients with COPD are frequently colonized with potential respiratory pathogens, and it is often difficult to identify conclusively a specific species of bacteria responsible for a particular clinical event. Bacteria frequently implicated in COPD exacerbations include Streptococcus pneumoniae, Haemophilus influenzae, and Moraxella catarrhalis. In addition, Mycoplasma pneumoniae or Chlamydia pneumoniae are found in 5–10% of exacerbations. The choice of antibiotic should be based on local patterns of antibiotic susceptibility of the above pathogens as well as the patient’s clinical condition. Most practitioners treat patients with moderate or severe exacerbations with antibiotics, even in the absence of data implicating a specific pathogen.
Glucocorticoids
Among patients admitted to the hospital, the use of glucocorticoids has been demonstrated to reduce the length of stay, hasten recovery, and reduce the chance of subsequent exacerbation or relapse for a period of up to 6 months. One study demonstrated that 2 weeks of glucocorticoid therapy produced benefit indistinguishable from 8 weeks of therapy. The GOLD guidelines recommend 30–40 mg of oral prednisolone or its equivalent for a period of 10–14 days. Hyperglycemia, particularly in patients with preexisting diagnosis of diabetes, is the most frequently reported acute complication of glucocorticoid treatment.
Oxygen
Supplemental O2 should be supplied to keep arterial saturations ≥90%. Hypoxemic respiratory drive plays a small role in patients with COPD. Studies have demonstrated that in patients with both acute and chronic hypercarbia, the administration of supplemental O2 does not reduce minute ventilation. It does, in some patients, result in modest increases in arterial PCO2, chiefly by altering ventilation-perfusion relationships within the lung. This should not deter practitioners from providing the oxygen needed to correct hypoxemia.
Mechanical Ventilatory Support
The initiation of noninvasive positive-pressure ventilation (NIPPV) in patients with respiratory failure, defined as PaCO2 >45 mmHg, results in a significant reduction in mortality rate, need for intubation, complications of therapy, and hospital length of stay. Contraindications to NIPPV include cardiovascular instability, impaired mental status or inability to cooperate, copious secretions or the inability to clear secretions, craniofacial abnormalities or trauma precluding effective fitting of mask, extreme obesity, or significant burns.
Invasive (conventional) mechanical ventilation via an endotracheal tube is indicated for patients with severe respiratory distress despite initial therapy, life-threatening hypoxemia, severe hypercarbia and/or acidosis, markedly impaired mental status, respiratory arrest, hemodynamic instability, or other complications. The goal of mechanical ventilation is to correct the aforementioned conditions. Factors to consider during mechanical ventilatory support include the need to provide sufficient expiratory time in patients with severe airflow obstruction and the presence of auto-PEEP (positive end-expiratory pressure), which can result in patients having to generate significant respiratory effort to trigger a breath during a demand mode of ventilation. The mortality rate of patients requiring mechanical ventilatory support is 17–30% for that particular hospitalization. For patients age >65 admitted to the intensive care unit for treatment, the mortality rate doubles over the next year to 60%, regardless of whether mechanical ventilation was required.
 _U.S. Dollar
_U.S. Dollar _Nepali Rupees
_Nepali Rupees